MONONEUROPATÍA
Dr. Guillemo Gualpa
MONONEUROPATÍA
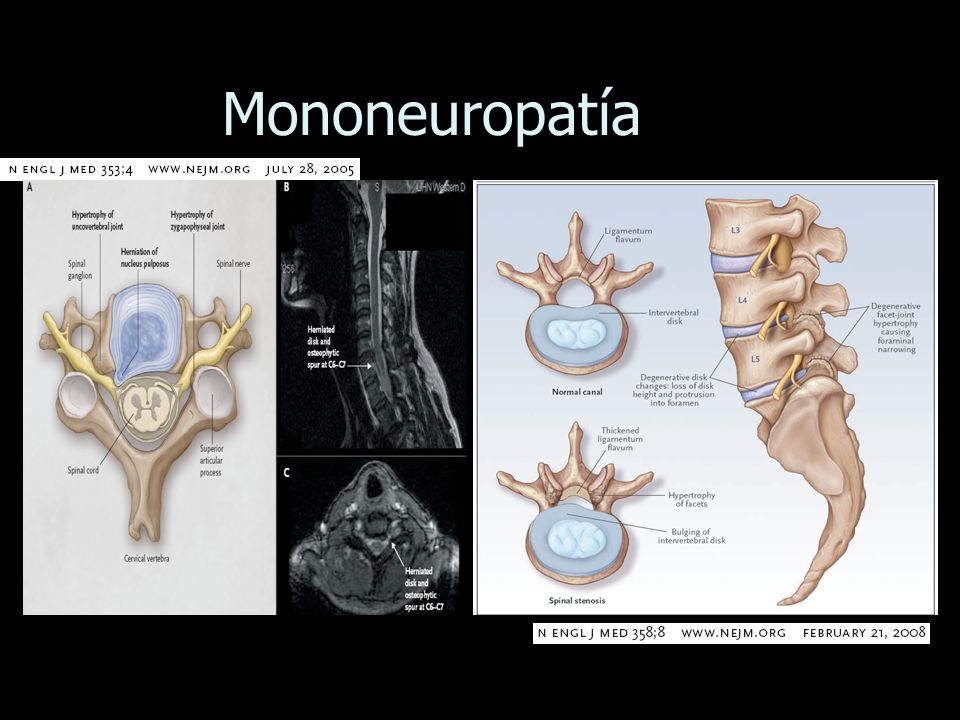
La mononeuropatía es una afección que provoca el daño de un solo nervio o grupo nervioso y perjudica la parte del cuerpo asociada con ese nervio o grupo nervioso, lo cual puede causar pérdida de la sensibilidad, del movimiento o de la función en esa parte del cuerpo. La mononeuropatía puede afectar cualquier parte del cuerpo.
Existen varios tipos de mononeuropatía, y su gravedad, frecuencia y síntomas dependen del tipo. Uno de los tipos más frecuentes es el síndrome del túnel carpiano, que se produce como consecuencia de la presión que se ejerce sobre el nervio mediano en el brazo, lo cual ocasiona entumecimiento, daño muscular y debilidad en las manos y los dedos. También existen varios tipos de mononeuropatía craneal que afectan los nervios del cráneo. La mononeuropatía del par craneal VI, por ejemplo, puede reducir la eficacia de los movimientos oculares y provocar visión doble.
Causas
La mononeuropatía es un tipo de daño a un nervio afuera del cerebro y la médula espinal (neuropatía periférica).
En la mayoría de los casos, la mononeuropatía es causada por lesión. Ocasionalmente los trastornos en todo el cuerpo (sistémicos) pueden causar daño a nervios aislados.
La presión prolongada sobre un nervio debido a hinchazón o lesión puede ocasionar mononeuropatía. La cubierta del nervio (vaina de la mielina) o una parte de la neurona (el axón) pueden resultar dañadas. Este daño retarda o impide que las señales viajen a través de los nervios afectados.
Tipos
Si los síntomas se presentan lentamente, la afección se denomina "neuropatía crónica"; si los síntomas se presentan de forma repentina, la afección se denomina "neuropatía aguda".
La neuropatía puede ser hereditaria. El tipo más frecuente de neuropatía hereditaria es la artropatía neuropática, que afecta la parte inferior de la pierna y el pie.
La neuropatía adquirida es mucho más frecuente y suele ser producto de una enfermedad o una lesión. Si las lesiones nerviosas son resultado de la diabetes, la afección se denomina "neuropatía diabética". Si se desconoce la causa, la enfermedad recibe el nombre de "neuropatía idiopática".
Síntomas
Los síntomas específicos dependen de los nervios afectados y, entre ellos, pueden incluirse los siguientes:
pérdida de la sensibilidad
hormigueo y ardor
falta de sensibilidad y entumecimiento
falta de coordinación
pérdida de los reflejos
fasciculación, calambres o espasmos musculares
debilidad
dolor
atrofia muscular progresiva
dificultad para moverse y parálisis
Diagnóstico
Si tiene síntomas de lesiones nerviosas, consulte al médico lo antes posible. Esté preparado para ofrecer detalles sobre sus antecedentes médicos completos y para informarle si toma algún medicamento o suplemento recetado o de venta libre.
El médico le realizará una exploración física completa y quizás solicite pruebas diagnósticas para determinar la causa de la afección; entre ellas, las siguientes:
electromiografía (EMG), que permite registrar la actividad eléctrica de los músculos
estudios de conducción nerviosa, que permiten registrar la velocidad de la actividad eléctrica de los nervios
biopsia del tejido nervioso, que consiste en extraer una porción pequeña de un nervio para ver si presenta daños.
Tratamiento
El tratamiento depende de la causa subyacente y de la gravedad de la lesión nerviosa. En algunos casos, la parte del cuerpo afectada quizás mejore sola, lo cual implica que no es necesario realizar tratamiento